 Leanpub
Leanpub
ANMs account for the direction of protein fluctuations
The generalization of a Gaussian network model, in which we attempt to determine the directions of the forces influencing alpha carbons, is called an anisotropic network model (ANM). Although ANMs include directionality, they typically perform worse than GNMs when benchmarked against experimental data1. However, ANMs can be used to create animations depicting the range of motions and fluctuations of the protein, as well as to estimate the specific directions of movement caused by each of a protein’s modes.
We will not delve into the mathematical intricacies of ANM calculations, but we will use ANMs to create animations visualizing protein fluctuations. For example, click on the animation below to see a video of estimated hemoglobin fluctuations produced from ANM. We can see that the left and right side of the protein are more flexible than the rest of the protein and twist in opposite directions.
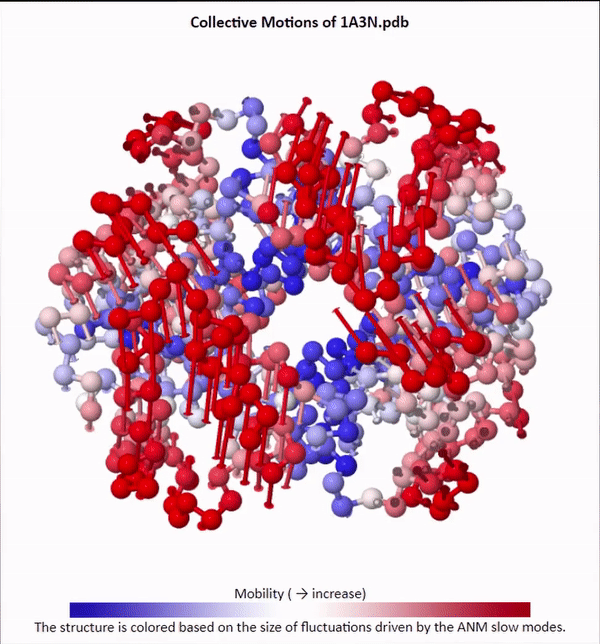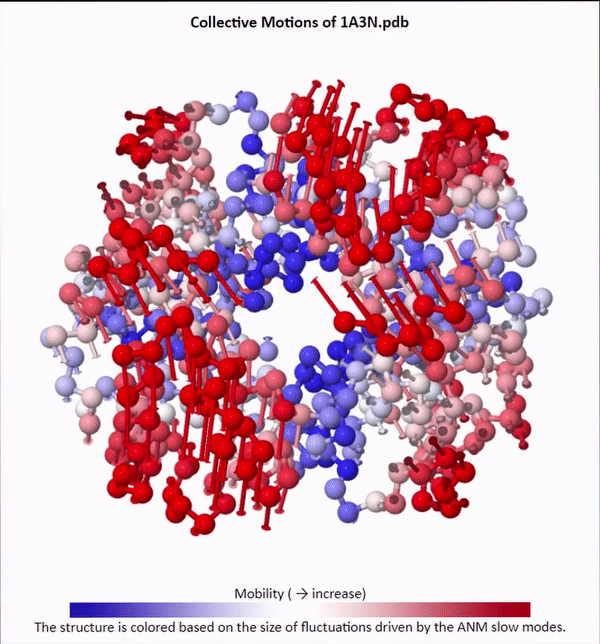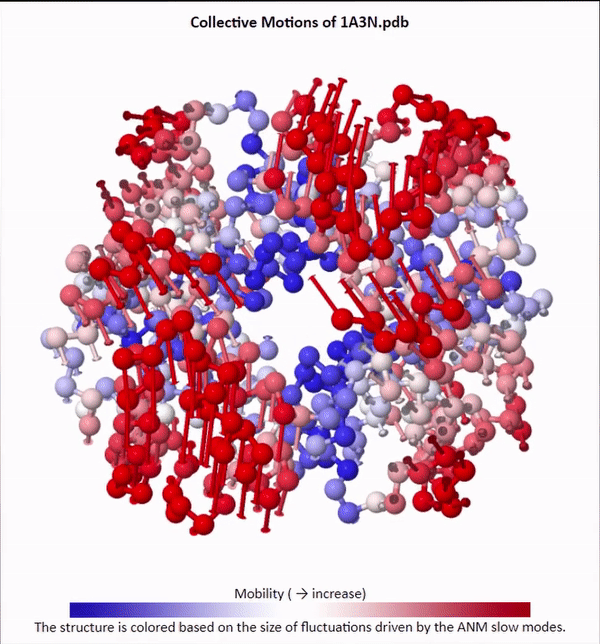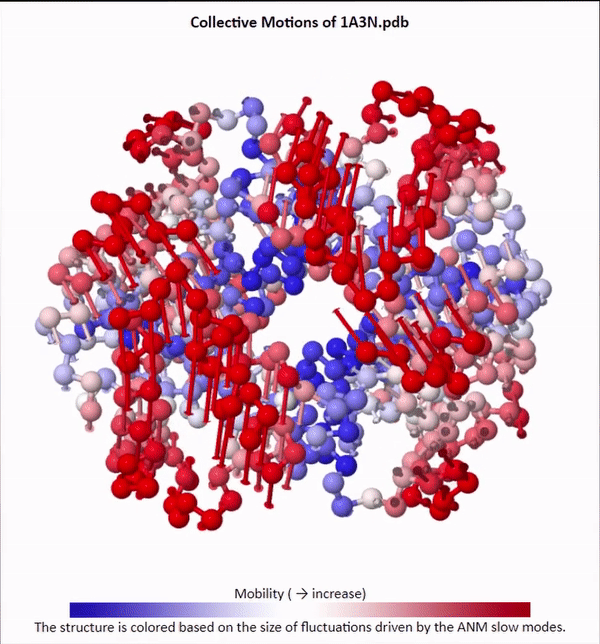 Collective motions of the slowest mode in human hemoglobin from ANM calculations using DynOmics.
Collective motions of the slowest mode in human hemoglobin from ANM calculations using DynOmics.
After we produce an animation like the one in the figure above, we also should attempt to explain it biologically. Human hemoglobin exists in two states: the tense state (T), in which it is not bound to an oxygen molecule, and the relaxed state (R), in which it is oxygenated. Hemoglobin’s mobility shown in the above animation corresponds to its ability to transition between these two states, in which salt-bridges and contacts can shift by up to seven angstroms2. This significant molecular flexibility exemplifies why we need to study protein dynamics as well as structure.
We will now apply ANM to the SARS-CoV and SARS-CoV-2 spike proteins. We will also use NMWiz, which is short for “normal mode wizard”, to perform ANM calculations and create an animation of the SARS-CoV-2 (chimeric) RBD and the SARS-CoV RBD.
Note: Although we have separated our discussion of GNM and ANM, the DynOmics 1.0 server is an effort to integrate these approaches on a user-friendly platform. If you are interested, an additional tutorial shows how to use DynOmics to replicate some of the analysis below.
ANM analysis of the coronavirus binding domain
The following two animations show the complex of each virus’s RBD (purple) bound with ACE2 (green). The following tables indicate the color-coding of each animation.
SARS-CoV spike protein RBD (PDB: 2ajf)
| SARS-CoV RBD | Purple |
|---|---|
| Resid 463 to 472 (Loop) | Yellow |
| Resid 442 (Hotspot 31) | Orange |
| Resid 487 (Hotspot 353) | Red |
| ACE2 | Green |
| Resid 79, 82, 83 (Loop) | Silver |
| Resid 31, 35 (Hotspot 31) | Orange |
| Resid 38, 353 (Hotspot 353) | Red |
SARS-CoV-2 spike protein chimeric RBD (PDB: 6vw1)
| SARS-CoV-2 (Chimeric) RBD | Purple |
|---|---|
| Resid 476 to 486 (Loop) | Yellow |
| Resid 455 (Hotspot 31) | Blue |
| Resid 493 (Hotspot 31) | Orange |
| Resid 501 (Hotspot 353) | Red |
| ACE2 | Green |
| Resid 79, 82, 83 (Loop) | Silver |
| Resid 31, 35 (Hotspot 31) | Orange |
| Resid 38, 353 (Hotspot 353) | Red |
Recall from the previous lesson that the greatest contribution of negative energy to the RBD/ACE2 complex in SARS-CoV-2 was the region called “hotspot 31”, which is highlighted in blue and orange in the above figures. If you look closely, as the protein swings in to bind with ACE2, the blue and orange regions appear to line up just a bit more naturally in the SARS-CoV-2 animation than in the SARS-CoV animation. That is, the improved binding that we hypothesized for a static structure appears to be confirmed by dynamics simulations. This animation provides yet one more piece of evidence that SARS-CoV-2 is more effective than SARS-CoV at binding to the ACE2 enzyme.
-
Yang, L., Song, G., Jernigan, R. 2009. Protein elastic network models and the ranges of cooperativity. PNAS 106(30), 12347-12352. https://doi.org/10.1073/pnas.0902159106 ↩
-
Davis, M., Tobi, D. 2014. Multiple Gaussian network modes alignments reveals dynamically variable regions: The hemoglobin case. Proteins: Structure, Function, and Bioinformatics, 82(9), 2097-2105. https://doi-org.cmu.idm.oclc.org/10.1002/prot.24565 ↩
